| Run eShadow |
| |
http://eshadow.dcode.org/
|
| Publication reference |
| |
I. Ovcharenko, D. Boffelli, and G. Loots, eShadow: A Tool for Comparing Closely Related Sequences, Genome Research, 14(6), 1191-1198 (2004)
|
| Description |
| |
This program is a tool for performing comparative sequence analysis of multiple closely-related nucleotide or protein sequences. Based on the assumption that mutations in different lineages accumulated independently of each other during evolution, eShadow analyzes ClustalW multiple sequence alignments of sequences indistinguishably similar in pairwise comparisons and identifies regions that accumulated small amounts of mutations throughout evolution. eShadow implements two different complimentary approaches: Hidden Markov Model Islands (HMMI) and Divergence Threshold (DT) to distinguish between functional vs neutrally evolving regions; detailed description for these methods can be accessed from the graphical output page.
eShadow is an automated tool with an internally incorporated ClustalW aligner. It does not require either an e-mail address or any private information for proper operation, instead it provides a user with a dynamically changing progress report of the analysis pipeline performance.`
|
| Input file |
| |
eShadow requires a set of orthologous sequences in the FASTA format. Every sequence should have a header line with the name of the species followed by nucleotide sequence consisting of [ACTGN] symbols in a format similar to this:
> Chimp
TTGGAGTGAGCAGTCAGAGCACAGTTTAGTGCAGCACATTGATAGGAA
ACTCTCTTGATGGTGGGGGAGATGAGAGAAAGCAAGAAAAGAAAGAAA
GGGGAAAGACTTGAGTGTGCTTAATGCT
> Bonobo
CAGTTTCGTGCAGCACACTGAGAGGAAGTGGAGACTACAAAGGCAGAC
AGAGAAAGCAAGAAAAGAAAGAAAGGAGCATAGGGGAGGGGCACAGGG
TGCTGGTGGAAGGAAGCAGGTGGAGAGTAAGGGAT
> Gorilla
CATTGATGAAAGCGTTTTCAGGAGATGGAGTGAGCAGTCAGAGCACAT
AGGCAGACAACTCTTGATGGTGAGGAAGATGAGAAAGCAAGAAAAGAA
CACAGGGG
|
Example multiple sequences FASTA file
|
| Details |
| |
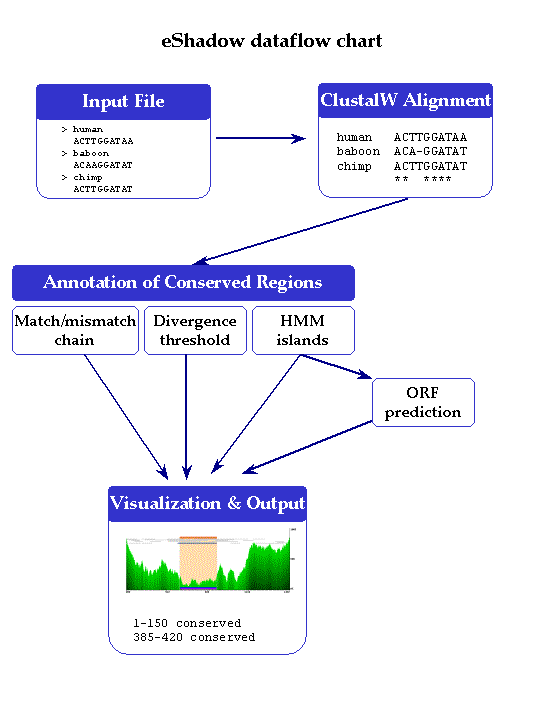 The overall scheme of eShadow dataflow is presented on the chart below. First, eShadow forwards the FASTA files submitted by the user to the locally installed ClustalW program. ClustalW multiple sequence alignments automatically distinguish between nucleotide and protein sequences, using different matrices to score different types of alignments. Repetitive elements are masked in the nucleotide sequences by the locally installed RepeatMasker and are forwarded together with the multiple sequence alignment to the visualization component.
The overall scheme of eShadow dataflow is presented on the chart below. First, eShadow forwards the FASTA files submitted by the user to the locally installed ClustalW program. ClustalW multiple sequence alignments automatically distinguish between nucleotide and protein sequences, using different matrices to score different types of alignments. Repetitive elements are masked in the nucleotide sequences by the locally installed RepeatMasker and are forwarded together with the multiple sequence alignment to the visualization component.
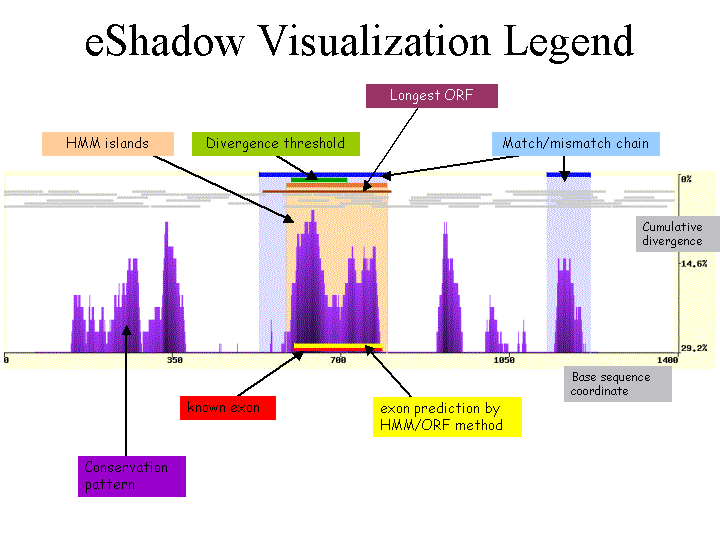
A distinct feature of the eShadow comparative sequence analysis tool is to extract functional elements based on the multiple sequence conservation pattern using two different approaches (Divergence threshold and Hidden Markov Model islands) taking into account the particular evolutionary history of the sequence under consideration. The extraction of conserved elements is augmented by the custom visualization plot (example picture) which dynamically adopts calculated conservation patterns and represents them in a set of differently colored bars overlaying the sequence conservation plot graph.
By overlaying Open Reading Frame (ORF) information on the predicted HMMI of conservation and requiring canonical AG-GT splice sites present at intron-exon boundaries to be conserved in all the species examined, eShadow is capable of detecting protein coding exons. The ORF detection feature is not intended to be used for de novo gene prediction, but as an indicator of putative protein character of a conserved segment.
Input sequences, ClustalW alignments and outputs, parameters for plotting evolutionary trees, and eShadow text outputs are available in addition to conservation plots. eShadow text outputs include basic statistics of the input data and multiple sequence alignment and provides with an analysis of the multiple sequence alignment (match/mismatch) pattern. Also it performs a significance analysis of different sequences, indicating which species introduces the main information content into the characterization of conserved elements in the multiple sequence alignment.
More details on data analysis and visual data representation are available at the eShadow help portal 
|
| Plot Legend |
| |
|
| Questions or suggestions? Contact us |
|
|
|

{kind=link}